MUsmanJaved007
Here is some information regarding your issues:
Larger system
You have several issues regarding your input structure:
- The coordinates are given in
atom.fracxyz while they are actually in cartesian coordinates. In fractional coordinates, the input values can only be between 0 and 1.
- Although you have a periodic system but you still included atoms on the boundary, so you have duplicated atoms in your structure.
- You set
domain.latvec = 4.54*[1 0 0; 0 1 0; 0 0 1]. For this unitcell atoms like 0.000000 4.546470 0.000000 lie out of the cell boundary.
So, in conclusion, your input structure does not represent a physically meaningful system.
Smaller system
Here you corrected the previous structure, but still there are few problems with your input parameters:
The system has a zero band-gap (metallic), and you need to use a non-zero smearing kpoint.sigma in order to converge the ground-state
Now, for a mettalic system, dielectric constant and born-effective charges are not well-defined. So it is not physically meaningful to simulate such ill-defined properties for metals. That's why you get the error (it's sort of a warning actually).
Also, in generall, you need to perform convergence test for the followng parameters, to make sure your final results are accurate enough
domain.lowres, current 0.5 value is very coarse
kpoint.gridn, for metals you probably need denser sampling. Please note that if you want to later perform a dfpt calculation, you need to use a gamma centered and odd grid.
kpoint.sigma
I encourage you to also consult RESCU's documentaion which include many of the points mentioned here. For dfpt calculations for metals please see this section
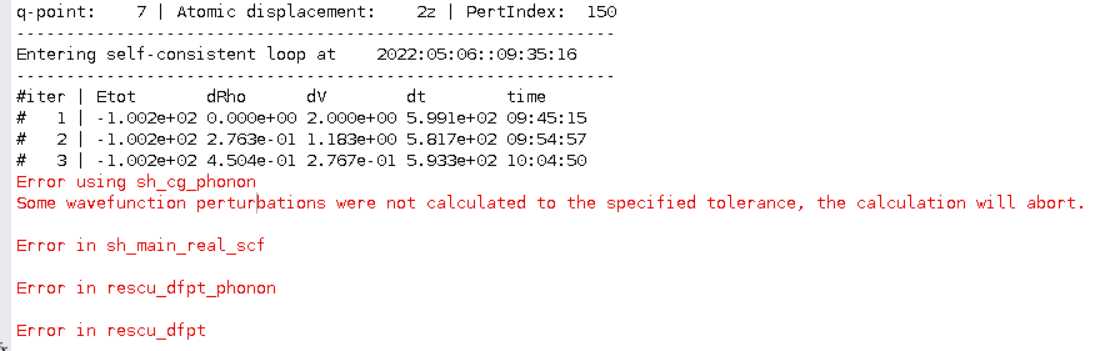
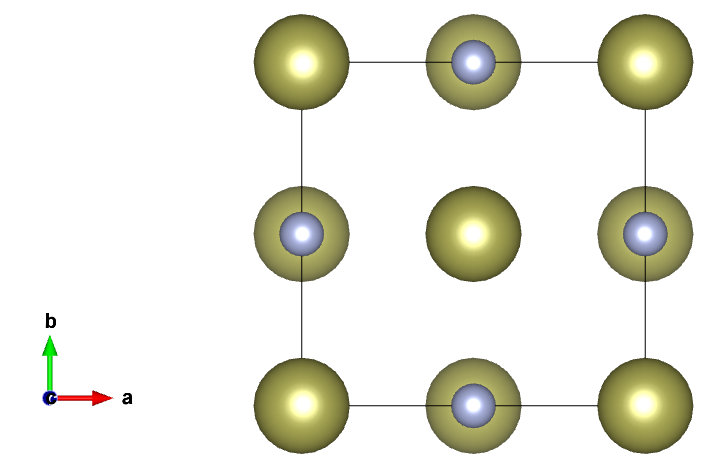
I was able to calculate phonons with the following input files:
scf.input
info.calculationType = 'self-consistent'
info.savepath = './results/HfN_scf';
atom.element = [1, 1, 1, 1, 2, 2, 2, 2]
atom.fracxyz = [0.0 0.0 0.0
0.0 0.5 0.5
0.5 0.0 0.5
0.5 0.5 0.0
0.5 0.0 0.0
0.5 0.5 0.5
0.0 0.0 0.5
0.0 0.5 0.0];
domain.latvec = 4.54*[1 0 0; 0 1 0; 0 0 1];
domain.lowres = 0.3;
element(1).species='Hf'
element.path = './Hf_TM_LDA.mat'
element(2).species='N'
element(2).path = './N_TM_LDA.mat'
units.length = 'a'
diffop.method = 'fft';
functional.list = {'XC_LDA_X','XC_LDA_C_PW'};
kpoint.gridn = [5,5,5]
kpoint.sigma = 0.002 % advised for dfpt
mixing.tol = 1e-5*[1 1] % advised for dfpt
option.saveWavefunction = 1 % required for dfpt
diffop.method = 'fft' % required for dfpt
domain.fourierInit = false % required for dfpt
phonon.input
info.calculationType = 'dfpt-phonon'
info.savepath = './results/HfN_phonon'
rho.in = './results/HfN_scf.mat'
psi.in = './results/HfN_scf.h5'
mixing.tol = 1e-5*[1 1] % advised for dfpt
option.saveWavefunction = 0
symmetry.spacesymmetry = 1
symmetry.pointsymmetry = 1
symmetry.timereversal = 1